


EPI2ME 24.08-01 Release
August 15, 2024
1 min
Dear EPI2ME Labs Users
With the June 2021 update to EPI2ME Labs, we are pleased to release a new workflow, an updated workflow and a comment on the re-invigoration of our tutorials.
wf-transcript-target is a our new Nextflow pipeline that reviews and consolidates transcripts of interest from direct RNA sequencing collections. The ambition of the workflow was originally to address the presence and fidelity of expected transcripts from in vitro transcription systems. A FASTQ format sequence collection is mapped to one or more transcripts of interest and summary information describing the mapping and coverage characteristics is collated. The starting sequence reads are also used to prepare a consensus sequence which is contrasted against the reference sequence.
This workflow can also be used for more general assessment of transcripts of interest. The figure below shows an excerpt of the HTML report produced when we look for the gusb ‘housekeeping’ gene in ENA sequence collection ERX4296980.
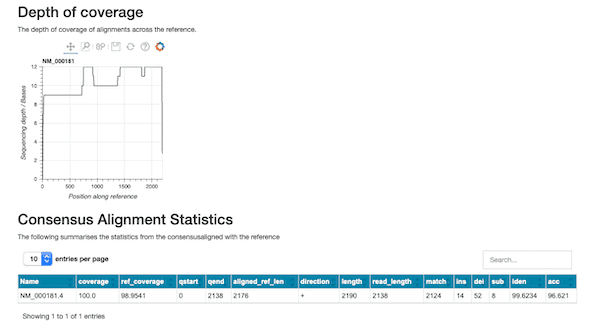
Our wf-metagenomics pipeline has been updated to collate additional taxonomic information from the Centrifuge analyses. This updated workflow will form the foundations for applied and focused metagenomic workflows that are in planning. The workflow has also been enhanced so that the Centrifuge HPVC indices (please see https://ccb.jhu.edu/software/centrifuge/ for more details) can optionally be downloaded and configured at runtime to simplify the analysis of broad metagenomic samples.
We are continuing to update and re-invigorate our EPI2ME Labs tutorials following last month’s update to Jupyter version 3. This housekeeping exercise is synchronising the usage of libraries and plots between the Nextflow workflows and the EPI2ME Labs tutorials. The SARS-CoV-2 ARTIC analysis tutorial now includes new functionality such as the Pangolin-based strain identification and re-establishes functionality parity with wf-artic, our Nextflow pipeline for SARS-CoV-2 genome analysis.
The medaka software has been updated to version 1.4.2 - the developments behind this update are associated with the calling and genotyping of genetic variants; please see the changelog for further details.
We look forwards to any feedback and would welcome recommendations for future content.