


EPI2ME 24.08-01 Release
August 15, 2024
1 min
Dear Nanopore Community,
Our (second!) July 2021 EPI2ME Labs release includes various updates to workflows and tutorials.
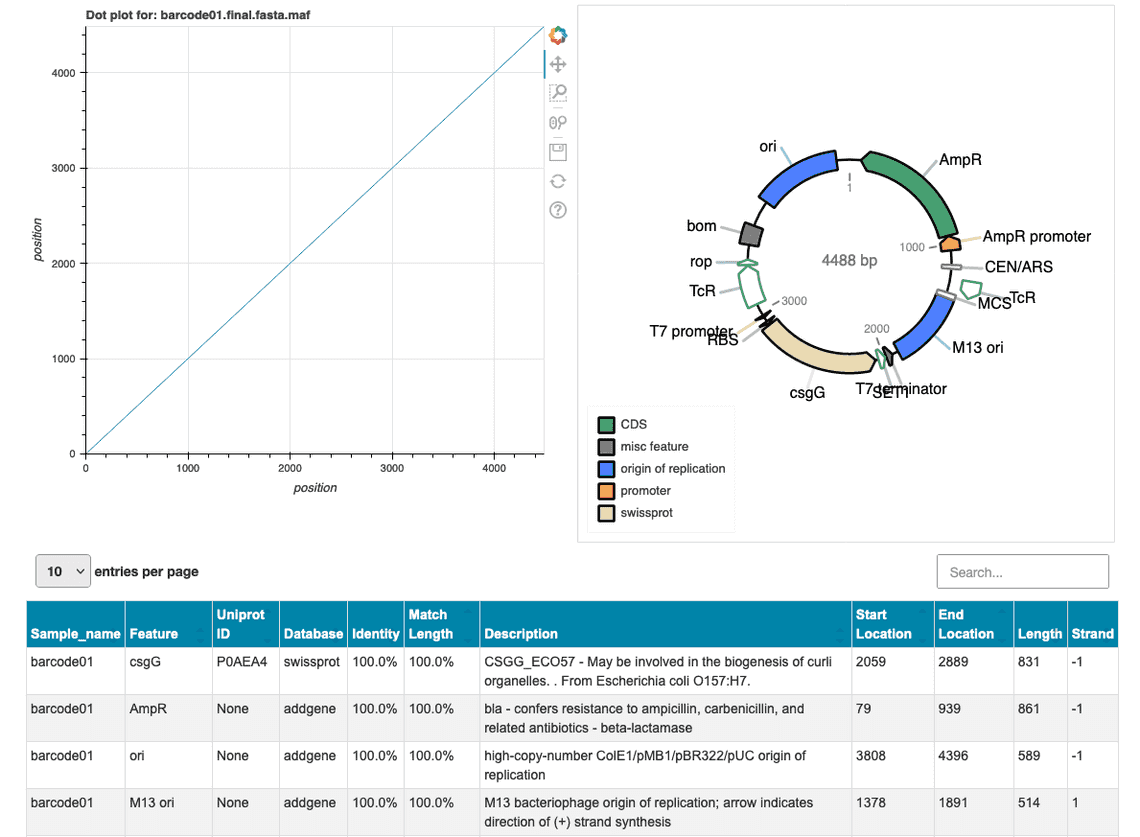
The wf-clone-validation workflow has been updated to version v0.1.0 and now includes the pLannotate tool for the visualisation and annotation of plasmid consensus sequences. The pLannotate software uses a collection of public sequence databases to explore the plasmid sequence for regions of interest that include e.g. the origin or replication and antibiotic resistance genes and may include information on the cloned insert sequence (if orthologues are present in the sequence database).
The wf-artic pipeline v0.3.1 release now reports a per-sample BAM file in the result directory. These BAM files are a standard component of the FieldBioinformatics workflow and correspond to the sorted sequence mapping data that has been structured by readgroup and trimmed of sequencing primers. This user request addresses the requirement that BAM files are also provided along with strain identification information.
The EPI2ME Labs SV tutorial has been updated to use LRA for read mapping and CuteSV for the identification of genomic structural variation; this synchronises the functionality between our research tool, pipeline-human-sv, and the EPI2ME Labs offerings. As a brief reminder, EPI2ME Labs provides a collection of best-practice tutorials that explore a variety of themes in the analysis of Nanopore sequence data. The EPI2ME Labs quick start guide provides more information.
The SARS-CoV-2 tutorial in EPI2ME Labs has been repaired - the V1200 primer set has been added back to the workflow.
We have made a number of minor changes to the way that we prepare and distribute our nextflow based workflows.
We look forwards to feedback and recommendations for future tutorials and workflows.